
A list of possible FDA reforms
Some of these are easier to get done than others...

Here’s a list of possible FDA reforms. I’m sure there are other ideas I’ve missed. If you have reform ideas you think should be added to this list please comment or DM me on Twitter.
I have ranked these according to my own perception of which have the most support among DC bureaucrats and members of congress.
Improving transparency. During the COVID-19 pandemic the public has been kept in the dark regarding how key decisions were made, such as the decision to issue an EUA for hydroxychloroquine, the decisions that pushed Pfizer's EUA from early November to late December, and the decision to require additional Phase III data from AstraZeneca which delayed their EUA by at least 3-4 months. Currently the FDA keeps much of the information relevant to decision making confidential. In theory Freedom of Information Act (FOIA) requests can be used to obtain some information, but in practice FOIAs are slow-walked, may require litigation, and take years to resolve. There are many possible ways to increase transparency at the FDA, some of which are outlined in this recent article in STAT News. 18 specific recommendations can be found in this journal article from 2018. One of people referenced as a top candidate for FDA Commissioner, Joshua M. Sharfstein, is a poor choice when it comes to implementing other reforms but has been a leading voice calling for more transparency.
Reciprocity. If a drug/medication is approved by a regulatory agency in a different country which has equivalent standards to the FDA (for instance agencies in the UK or Japan, the European Medicines Agency (EMA), or Health Canada), it should automatically be approved by the FDA (and vice versa). Reciprocity would save both taxpayers and companies a lot of time and money. Imagine if the AstraZeneca vaccine had been approved on January 1st, a few days after the UK approved it. It’s easy to see that tens of thousands of lives would have been saved, especially when you consider it would have been given to the most at-risk first! In 2015 Senators Mike Lee and Ted Cruz introduced the RESULT Act, which is primarily focused on implementing reciprocity.
Making the agency independent from the executive branch. It’s hard to insulate the FDA from political concerns as long as Congress controls the FDA’s purse, but it could at least be removed from direct interference from the executive branch by making it an independent agency like the FCC or Federal Reserve. There seems to be wide support behind this idea right now and recently four former FDA commissioners all endorsed this idea in an interview.
Rolling reviews. It should not take 3-4 weeks from the submission of an EUA application until a decision is made, especially when thousands are dying every day while waiting for vaccines. According to Dr. Marty Makary, a professor of public health policy at the Johns Hopkins Bloomberg School of Public Health who has conducted over a hundred clinical studies, the FDA “could have done the approval in 24-48 hours without cutting any corners”. Likewise, the approval of drugs and therapies after Phase III trials have reached an endpoint should not take 6 - 18 months. While the FDA does engage in a back and forth with companies prior to when they submit their paperwork for an EUA or approval, they do not use rolling reviews. Due to their rolling review systems other countries like the UK were able issue EUAs for COVID-19 vaccines faster than the FDA. Rolling reviews should be the norm, and the submission and analysis of trial data should made as streamlined and as efficient as possible without compromising the integrity of the analysis.
Tiered approvals. Doctors who want to provide new drugs to at-risk patients currently have to wait 5-10 years for lengthy Phase III trials to conclude and then another 6 - 18 months for the FDA to carry out their review of the trial data. Tiered approvals would allow a lower level of approval after just Phases I and II, freeing up treatments to those who need them most. The centrist Niskanen center has a white paper which suggests four levels of approval (see also their op-ed in The Hill).
Expansion of Right-to-Try. Federal right-to-try legislation was passed in 2018. However, it is very restrictive, and patients need to have met a number of strict requirements before they can try new medications and treatments, greatly limiting its utility.
Treating aging as a disease. Currently it is illegal to market an FDA-approved product as a treatment for aging. Even though aging is a harmful biological process, it is not considered a legitimate “indication” for a drug or therapy by the FDA. In other words, the FDA doesn’t view aging as a “disease” and therefore anti-aging treatments fall outside their mandate. Some specific aspects of aging are also not considered as legitimate indications. The FDA is currently operating in an inconsistent way as some conditions which are due to the aging process can are considered legitimate targets, such as osteoporosis and menopause, but others, like sarcopenia (age-related muscle loss /frailty), are not. Many experts, including David Sinclair, have spoken out about this issue. Some companies have found ways to get around the FDA's restrictions, such as by using certain metabolic markers to track the degree of damage from aging. However, the impossibility of getting FDA approval for therapies that directly slow down or repair the damages from aging greatly dis-incentivizes industry R&D investment in this area. Fortunately, with advanced gene and stem-cell therapies on the horizon, the Congress and the FDA have already taken a few steps towards being able to review and approve anti-aging drugs and therapies. The 21st-Century CURES Act, for instance, mandated the creation of the Regenerative Medicine Advanced Therapy designation at the FDA.
Challenge trials in emergency situations. Many people, including a group of legislators, lobbied the FDA to give companies the go-ahead to do challenge trials, but there was no action. Thousands of volunteers for COVID-19 challenge trials signed up with 1daysooner.org but were unable to participate as they had wished to. As a result, data on vaccine efficacy was obtained much slower than it could have been.
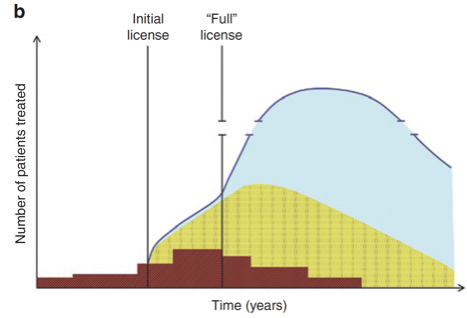
Adaptive licensing. Many different variations on this are possible, and figuring out the optimal procedure for a given candidate treatment is complex. However, the basic concept is quite simple — instead of waiting the normal 1-5 years for Phase III trials to conclude, you grant an “initial approval” based on interim data during the trial. Besides making promising treatments available to at-need patients sooner, another benefit is you can start gathering observational data in addition to the RCT data, a process which is relatively cheap and compared to the RCT. The process is summarized in the following two figures:


Figures from Eichler et al. Clinical Pharmacology & Therapeutics, 91: 426-437: Use of the proactionary principle for all drug & therapy approvals. The FDA should publish a transparent, quantitative, scientifically informed, and structured cost-benefit analysis for each regulatory action performed, which estimates the expected quality-adjusted life years (QALYs) saved versus risk of QALYs lost for both the action and inaction. The analysis should be made public, ideally sometime before the decision goes into effect. Crucially, the analysis should enumerate the risks and benefits of granting an approval and the risks and benefits of not granting it. See Max More’s overview of the proactionary principle and his chapter in The Transhumanist Reader, where he presents not just a principle but an entire framework for rational decision making. The key reform here is to make it mandatory that decision making at the FDA be highly structured and quantitative so it under less sway from political concerns and cognitive biases. If such a framework for rational decision making was in place it’s unlikely the FDA would have decided to delay Pfizer’s Emergency Use Authorization (EUA) from early November to December 11th, a decision which cost tens of thousands of American lives.
Free to Choose Medicine with a Trade-off Evaluation Database. In brief, Free to Choose Medicine would create an additional track after Phases I and II to allow doctors to prescribe new therapies to at-risk patients who are unable or unwilling to participate in a Phase III trial. Patients in the Free-to-Choose Track would be mandated to submit data to a trade-off evaluation database, creating a trove of valuable real-world data (this could be genetic, biomarker, and adverse events data, for instance). See this excellent podcast interview with Bart Madden for more information.


Great post!
Implementing any of these would prevent millions of life years of suffering.
Thank you for your service to humanity 🫡